









Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.19 n.2 Caracas jul. 2000
Heparinas de Bajo Peso Molecular en la Cardiopatía Isquémica Metabólica. Bases Moleculares. Experiencia Clínica y Resultados
F Bermúdez A1, V Bermúdez P1, C Cano P1, M Medina R1, M Núñez P1, H Restrepo1, ME Vargas1, D García1, A Souki1, H Seyfi1 y A Pérez L1.
- Centro de Investigaciones Endocrino Metabólicas "Dr Félix Gómez" Facultad de Medicina. Universidad del Zulia, Maracaibo.
RESUMEN
Las Heparinas de Bajo Peso Molecular (HBPM) se aíslan a partir de la heparina no fraccionada, siendo su mecanismo de acción el efecto inhibitorio que ejerce sobre el factor Xa por la antitrombina. Las HBPM más utilizadas a nivel mundial son la enoxaparina (Clexane), nadroparina (Fraxiparina) y la dalteparina sódica (Fragmin). Un considerable número de ensayos clínicos ha comparado el efecto de las HBPM con la NHF en el tratamiento de la trombosis venosa profunda y del tromboembolismo pulmonar donde se confirma la eficacia de las HBPM sobre las NHF. Con base a estudios anteriores, utilizamos la dalteparina sódica a dosis de 5000 UI/día durante 10 días; indofubeno a dosis de 400 mg/día y trimetazidina a dosis 60 mg/día durante 3 meses en pacientes con cardiopatía isquémico - metabólica. Nuestros resultados muestran una mejoría significativa en la mayoría de los pacientes observándose estabilización en la frecuencia cardíaca , desaparición del dolor precordial y desaparición de los signos de isquemia, lesión y necrosis en el electrocardiograma, por lo que sugerimos la utilización de esta terapéutica no invasiva y ambulatoria en pacientes con cardiopatía Isquémico - metabólica.
Palabras Claves: Heparinas de Bajo peso molecular, Heparina no fraccionada, Antitrombina, Cardiopatía Isquémico metabólica, Dalteparina sódica.
ABSTRACT
Low molecular weight heparin (LMHW) is isolated from unfractionated heparin (UFH). LMWH mechanism of action is exerted on Factor Xa through antithrombin. The most used LMWH are enoxaparin (Clexane), nadroparin (Fraxiparin) and deltaparin (Fragmin). A significant number of clinical assays have compared the effects of LMWH and UFH on deep venous thrombosis and pulmonary thromboembolism, showing LMWH better results than UFH. Based on these studies, dalteparin was used at a dose of 5000 IU/ day during 10 days; indobuphen 400 mg/day and trimetazidine 60 mg/day during 3 months in patients with ischemic cardiopathy. Our result show a significant improvement in these patients, through heart rate stabilization and absence of chest pain as well as tissue damage and necrosis in EKG. According to these results we suggest the use of this non invasive and ambulatory therapy in patients with ischemic cardiopathy.
Key Words: Low Molecular Weight heparin, Unfractionated heparin, Antithrombin, Ischemic-metabolic cardiopathy, Dalteparin.
BASES MOLECULARES
La oclusión trombótica de arterias y venas da cuenta de aproximadamente del 60% de todas las causas de mayor morbilidad y mortalidad en pacientes hospitalizados y es responsable de la mayoría de los síndromes agudos, subagudos y crónicos coronarios (1,2). Existen tres eventos importantes en la patogénesis de la trombosis arterial: 1. La placa aterosclerótica ocluye progresivamente el vaso creando flujo turbulento, aunque generalmente no llega a la oclusión por sí sólo; 2. La placa inestable (pequeña, con una cubierta fina y un gran corazón lipídico) se rompe, exponiendo el tejido subendotelial a la circulación y en consecuencia a las substancias protrombóticas; 3. En respuesta a la ruptura de la placa, las plaquetas se adhieren y se agregan, generando trombina y finalmente fibrina lo que conduce la aparición de un trombo en muchas ocasiones de tipo oclusivo (1).
Hace mas de 15 años la atención de los clínicos fue puesta en el manejo de la isquemia miocárdica con drogas antiplaquetarias, que aún se mantiene, aunque la evidencia actual está muy a favor de que la inhibición de la actividad de la trombina es de particular importancia a este nivel (3,4). La función principal de la trombina es la de catalizar la conversión del fibrinógeno a fibrina. Una vez que la trombina se ha formado es capaz de unirse a la fibrina, y aún así permanece activa, pudiendo inclusive amplificar su propia generación de 300 a 400 veces más que mediante los factores V y VII. Además de todo esto, la trombina es un potente activador de plaquetas. Al hacer un análisis detallado de los procesos mencionados, no es raro pensar porqué la inhibición de la actividad trombínica es una de las tantas formas de manejar adecuadamente la cardiopatía isquémica metabólica.
La trombina puede inhibirse por numerosas y diversas maniobras: 1. Mediante la administración de inhibidores directos de su actividad (hirudina, hirulog e hirugen). 2. Mediante la administración de inhibidores dependientes de la actividad de la antitrombina III (heparina no fraccionada y heparinas de bajo peso molecular). 3. Por la acción de inhibidores indirectos (Warfarina).
Inhibidores directos de la actividad trombínica (hirudina, hirulog, hirugen)
Existen dos tipos de inhibidores directos de la trombina, los naturales, como la hirudina (extraída de la saliva de la sanguijuela) y los sintéticos, obtenidos de la manipulación molecular del anterior (5,6). Ambos grupos inhiben la unión de la trombina con la fibrina, la cual en realidad es inaccesible a los inhibidores dependientes del efecto de la antitrombina III. Esta propiedad constituye un hito farmacológico en el sentido que el trombo residual actúa como un potente estímulo trombogénico. Esta diferencia entre los inhibidores directos y los inhibidores dependientes de la AT-III se relaciona con la forma de unión a la trombina. La trombina tiene tres sitios de unión: 1. Un sitio de unión a la fibrina. 2. Un sitio catalítico. 3. Un sitio de reconocimiento del substrato. Además de éstos, hay un sitio de unión a la heparina que se encuentra muy cerca al sitio de unión a la fibrina. Una vez que la trombina se une a la fibrina ocurre un cambio conformacional que hace inaccesible al complejo ATIII-heparina (1).
Señalando un marcado contraste, los inhibidores directos de la trombina no requieren el acceso al sitio de unión de la heparina por lo que es un potente inhibidor de la actividad trombínica tanto de la que se encuentra unida a la trombina como la que se encuentra en fase fluida. El uso de los inhibidores directos de la trombina ha sido evaluado en forma extensa en diversas situaciones clínicas como en la angina inestable(7), infarto agudo del miocardio, angioplastia(8) y tromboembolismo venoso. Sin embargo, en 1994, tres estudios clínicos en fase III con hirudina tuvieron que ser descontinuados debido a sangrado excesivo. Es de particular interés el incremento del riesgo de sangrado intracraneano, particularmente cuando este fármaco se usa en combinación con aspirina como adyuvante de la trombolisis(9). Sobre la base de los resultados de todos estos estudios se puede concluir que el margen entre el efecto terapéutico y el efecto tóxico es muy bajo.
Heparina no fraccionada
La heparina es un glucosaminoglicano que se encuentra en los gránulos secretores de las células cebadas mucosas y epiteliales (10). Se sintetiza a partir de carbohidratos activados con el UDP (uridin-difosfato), como un polímero de residuos alterados de ácido-D-glucurónido y N-acetil-glucosamina. En su forma natural forma parte de una gran proteína llamado proteoglicano, formado por una base proteica a la cual se unen 10 a 15 cadenas de glucosaminoglicanos (heparina) con una extensión de 200-300 unidades monosacáridas(11). Posteriormente los glucosaminoglicanos pueden sufrir modificaciones que incluyen la N-desacetilación y N-sulfatación de residuos de glucosamina, epimerización del ácido glucurónico a ácido L-idurónico, la O-sulfatación de residuos de ácido de residuos de ácido idurónico y glucurónico en posición C2 y la O-sulfatación de residuos de glucosamina en posición C3 y C6. Cada una de esas reacciones de modificación es incompleta y da origen a estructuras de oligosacárdios después que el proteoglicano heparina se ha transportado hacia el gránulo de secreción de la célula cebada. Entonces una enzima llamada endo-beta-D-glucuronidasa desintegra las cadenas de glucosaminoglicanos hacia fragmentos de 5.000-30.000 daltones durante un período de horas(11).
Figura 1: Parte Activa de la Heparina: Secuencia Pentasacárida de la Unión Antitrombínica de la Heparina
Figura 2: Principal Mecanismo de Acción de la Heparina no Fraccionada (HNF)
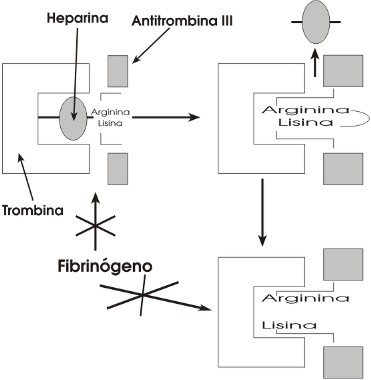
Mecanismo de acción
En 1939, Brinkhous y cols. describieron que el efecto anticoagulante de la heparina estaba mediado por un componente endógeno del plasma denominado "cofactor de heparina". Luego de 30 años se purificó la antitrombina III (ATIII) demostrándose que tenía la famosa actividad de cofactor de heparina (11). La ATIII es un polipéptido de una sola cadena con un PM de 58kDa que inhibe con rapidez a la trombina en especial en presencia de heparina. Esta proteína pertenece a la familia de los inhibidores de la alfa-1 antitripsina y es producida en el hígado y exportada a la circulación donde circula a una concentración de 2 _M, inhibiendo a los factores de coagulación activados IIa (trombina), Xa, IXa, XIa, XIIa y calicreína (vía común e intrínseca) (10). La ATIII es un substrato suicida para estos factores, ya que en realidad todos son proteasas. La inhibición ocurre cuando el factor de coagulación activado ataca el enlace Arginina-serina dentro de la ATIII, quedando atrapada en un complejo inhibidor 1:1 estable. La heparina aumenta alrededor de 1.000 veces la tasa de reacción entre la trombina y la ATIII al servir como una plantilla catalítica a la cual se unen tanto el inhibidor como la proteasa. La unión de la heparina también produce un cambio conformacional en la ATIII que hace que el sitio reactivo esté más accesible al factor de coagulación. Una vez que la trombina se une a la ATIII se libera la molécula de heparina del complejo. El sitio de unión de la heparina para la ATIII es una secuencia pentasacárida que se encuentra sólo en el 33% de la molécula de heparina(12).
La heparina no fraccionada (HNF), extraída de fuentes porcinas o bovinas ha estado disponible para el uso clínico durante varios decenios. Aún cuando la HNF se ha estudiado de manera extensa, persisten muchas dudas sobre su mecanismo de acción, en especial, las propiedades no anticoagulantes(4). No obstante, cada día se comprenden mejor algunas de las complicaciones asociadas al tratamiento heparínico(13).
Se ha demostrado que las moléculas de heparina formadas por menos de 18 unidades monosacáridas carecen de capacidad de unirse a la trombina y a la ATIII de manera simultánea, y ello da como resultado la imposibilidad de que catalicen la inhibición de la trombina, razón por la cual las heparinas de bajo peso molecular tiene un efecto menor contra la trombina y por ende, un menor poder anticoagulante.
La heparina también cataliza la inhibición de la trombina por otro cofactor plasmático (cofactor II) que actúa de modo independiente de la ATIII(11). La heparina tiene otros efectos sobre la hemostasia entre los que se incluye la inhibición de la liberación de factor tisular, la supresión de la función plaquetaria, la unión a proteínas plasmáticas y el aumento de la permeabilidad vascular(4).
A manera de resumen se puede concluir que la HNF tiene cinco efectos sobre la hemostasia: 1. Se combina con la AT-III para inhibir la coagulación. 2. A dosis altas se puede unir al cofactor de heparina II para inhibir directamente a la trombina. 3. Interfiere con la interacción de los factores de la coagulación entre sí en la superficie de la plaqueta. 4. Inhibe de manera directa la agregación plaquetaria y 5. Incrementa la permeabilidad vascular. Estos cinco efectos aumentan la posibilidad de fenómenos hemorrágicos, mientras que la inhibición de la cascada de la coagulación es la responsable principal de su efecto antitrombótico.
La HNF se usa ampliamente en la prevención y tratamiento del tromboembolismo venoso y en el manejo de la cardiopatía isquémica metabólica. La mayor parte del conocimiento clínico actual sobre el uso de la heparina en la angina inestable proviene de los estudios de Théroux y cols, en Montreal(14). En pacientes con angina inestable estos autores compararon la acción de la aspirina sola y la heparina IV. Se encontró que los pacientes que recibieron heparina resultaron con una reducción del 78% en el riesgo de infarto agudo del miocardio en comparación con el grupo que recibió aspirina(14). En un estudio posterior del Instituto del Corazón de Montreal se descubrió un efecto aditivo cuando se usaba la combinación de aspirina con heparina. Lamentablemente, la administración de heparina (12) se relaciona con un aumento en el riesgo de sangrado no deseado, trombocitopenia y la necesidad de monitorizar frecuentemente el tiempo parcial de trombina (TPT) (15), lo cual muchas veces requiere de hospitalización.
Heparinas de bajo peso molecular (HBPM)
La HNF es una mezcla de moléculas de glucosaminoglicanos con pesos moleculares de 2.000 a 30.000 daltones (promedio: 15.000 d). Desde un punto de vista químico, esta heparina puede fraccionarse mediante métodos químicos o físicos. Las moléculas de heparina con pesos moleculares entre 4.000 y 6500 d reciben el nombre de heparinas de bajo peso molecular (HBPM)(13). Esta fracción tiene efecto inhibitorio sobre los factores de coagulación activados, aunque con menor efecto potencial de sangrado cuando se compara con la HNF(13).
Tal vez la diferencia mas evidente entre la HNF y las HBPM es que éstas últimas tienen muy poco efecto sobre los resultados que arrojan las pruebas de coagulación. La HNF a baja concentración puede volver incoagulable la sangre, mientras que con las HBPM se requiere una dosis hasta 15 veces más de la usual para producir solo modestos cambios en el tiempo de protrombina (TP)(16) y TPT. Este hecho significa que bajo el tratamiento estándar con HBPM no se van a observar cambios en las pruebas de coagulación mientras que se mantiene su capacidad antitrombótica(17).
La corta extensión polisacárida de las HBPM hace que su interacción con el complejo Trombina-ATIII sea de menor afinidad y cuantía. Sin embargo, las HBPM mantienen la capacidad de catalizar la inhibición del factor Xa por la antitrombina III. De esta forma, a diferencia de la heparina no fraccionada, que tiene una relación anti-Xa/anti-IIa de 1:1, las HBPM poseen relaciones de 2:1 hasta 4:1(13).
Otras propiedades que distinguen a las HBPM incluyen:
1. Imposibilidad de ser inactivadas por el factor plaquetario IV, el cual sí es un potente inhibidor de la heparina durante la coagulación.
2. Inhibición de la actividad del factor Xa incluso cuando se encuentra en la superficie de las plaquetas formando el complejo protrombinasa. A este efecto se le atribuye la cualidad de evitar la activación de la protrombina en la superficie de la plaqueta y el bloqueo de la agregación plaquetaria mediada por trombina.
3. Incapacidad de unirse a proteínas ricas en histidina, fibronectina, vibronectina y al factor de von Willebrand a diferencia de la heparina, hecho que parcialmente neutraliza el efecto de esta última (heparina) sobre la inhibición del factor Xa.
Otra gran ventaja de las HBPM respecto a la HNF se deriva de sus diferencias en la depuración plasmática(18,19). La heparina no fraccionada es eliminada de la circulación mediante un mecanismo saturable relacionado con su unión a proteínas plasmáticas y al endotelio, recalcando, por supuesto, que la heparina unida a estos elementos carece de actividad biológica(20). En contrate con lo anterior, las HBPM no se unen al endotelio ni a proteínas plasmáticas, por lo que tiene una muy buena biodisponibilidad cuando se inyecta por vía subcutánea (21). Como resultado de esto las HBPM tienen una dosis/respuesta estable que mejora su utilidad clínica en comparación con la HNF(22,23).
Tabla 1: Algunas Heparinas de bajo peso molecular disponibles en el mercado y algunas de
sus propiedades Farmacocinéticas
| Nombre Comercial | Nombre genérico Internacional | Método de producción | Peso molecular medio | Proporción anti-Xa/anti-IIa | Vida Media (minutos) |
| LOGIPARIN | TINZAPARINA | DH | 5.866 | 1,9:1 | - |
| FRAGMÍN | DALTEPARINA | DAN | 5.819 | 2,1:1 | 119 |
| CLEXANE | ENOXAPARINA | HA | 4.371 | 2,7:1 | 129 |
| FRAXIPARINA | NADROPARINA | DAN | 4.855 | 3,2:1 | 129 |
| REVIPARINA | CLIVARINA | DAN | 4.653 | 3,6-6,1:1 | - |
| NORMOFLO | ARDEPARINA | DP | 6.000 | 2:1 | - |
| SANDOPARIN | CERTROPARIN | HA | 4.500 | 2:1 | 270 |
| FLAXUM | PARNAPARIN | HA | - | - | - |
DH= digestión con heparinasa, DAN= despolimerización con ácido nitroso, HA= hidrólisis alcalina, DP= Desdoblamiento peroxidativo.
Tabla 2: Comparación Química Entre la HNF y las HBPM.
| HBPM | Heparina no fraccionada | |
| Peso molecular medio | 4.000 – 6.500 | 12.000 – 15.000 |
| Unidades monosacáridas | 13 a 22 | 40 a 50 |
| Proporción de actividad anti-Xa/anti-IIa | De 2:1 a 4:1 | 1:1 |
| Biodisponibilidad a dosis bajas | Alta | Baja |
| Depuración dependiente de la dosis | No | Sí |
| Inhibición por factor plaquetario IV | No | ++ |
| Inhibición del factor Xa unido a plaquetas | Sí | No |
| Inhibición de la función plaquetaria | + | ++++ |
| Incremento de la permeabilidad vascular | No | + |
Tabla 3: HBPM, Algunas Ventajas en el Tratamiento de Condiciones que Cursan con Trombosis
| 1. | Dosis respuesta predecible |
| 2. | Vida media relativamente larga |
| 3. | Sangrado reducido o ausente para un efecto trombótico dado |
| 4. | Seguridad y eficacia en el tratamiento de pacientes con trombosis venosa o cardiopatía isquémica. |
| 5. | Uso ambulatorio. |
| 6. | Cómoda presentación y fácil aplicación. |
EXPERIENCIA CLINICA
Un considerable número de ensayos clínicos ha comparado el efecto de las heparinas de bajo peso molecular (HBPM) con la heparina no fraccionada (HNF) en el tratamiento de la trombosis venosa profunda y del tromboembolismo pulmonar consecutivo, en algunas personas que sufrieron estas dos enfermedades o una de ellas como consecuencia de un reposo prolongado en cama por enfermedad médica o después de un acto quirúrgico con alto riesgo, frecuentemente en el área abdominal, pelviana, de miembros inferiores o cardíaca. Se confirmó en ellos que la eficacia de las HBPM es similar o mejor que la HNF, conclusión a la cual llegaron por la disminución de la mortalidad (50 por ciento) y por el menor número de sangrados. Por otra parte, gracias a que no amerita monitoreo de laboratorio ni hospitalización, su índice costo/beneficio se reduce substancialmente.(24) Aunque los primeros estudios clínicos son relativamente recientes en cuanto al tópico venoso y pulmonar, últimamente se ha avanzado mucho en el área de la cardiopatía isquémica metabólica (33,34,35,36,37). En la tabla 2 se comparan los efectos de la HNF y la HBPM.
Tres de las HBPM más utilizadas mundialmente están disponibles en nuestro país. Ellas son la enoxaparina (Clexane), cuya presentación es de 4.000 y 6.000 UI, la nadroparina (Fraxiparina), en presentación de 3.000 UI (7.500 unidades Choay) y la dalteparina sódica (Fragmín), que se presenta en ampollas de 2.500, 5.000 y 10.000 UI(25,26,27). Todas las presentaciones se ofrecen en jeringas prellenadas. La técnica para su correcta administración son las siguientes: 1. Antes de quitar la funda protectora compruebe si la dosis contenida es correcta. 2. No es preciso purgar la jeringa para extraer la burbuja que se ve en ella. No se amerita hacerlo, porque esta burbuja es inofensiva y además no pasará al cuerpo. Si lo hace perderá una importante cantidad del contenido. 3. La zona de inyección es el área abdominal, a uno u otro lado del ombligo. 4. En posición recostada desinfectar la zona con alcohol y tomar con los dedos un pliegue cutáneo. 5. Sin soltar el pliegue introduzca la aguja perpendicularmente a la piel y empuje el contenido lentamente. 6. No frotar la zona después de la inyección.
Como la HNF, la HBPM actúa contra el factor Xa, antifactor que se ha relacionado con la liberación, por parte del hígado, de la lipasa trigliceridasa (HTGL) o lipoproteín lipasa(40), a la cual se debe principalmente su acción anticoagulante, hecho que motivó su empleo como profiláctico del tromboembolismo pulmonar. El efecto molecular de la heparina se debe a una competición de ella en la unión entre el heparán sulfato con la lipoproteín lipasa. Al hacerlo queda libre la lipoproteín lipasa, que entonces degrada en el plasma las moléculas de VLDL, aclarándolo. Por eso se le conoce como "aclarador del plasma turbio". Sin embargo, al escindir las moléculas de VLDL quedan libres los ácidos grasos unidos a la albúmina y el glicerol. Los primeros, es decir, los ácidos grasos son perjudiciales por aumentar la resistencia a la insulina. Pero además, las HBPM tiene otros efectos conocidos, entre los cuales se encuentran los ya mencionados en la revisión molecular. En la práctica clínica se ha observado por estudios diversos que las HBPM tienen acción anticoagulante, especialmente sobre el trombo rojo, similar a la HNF mediante su efecto sobre el factor Xa, y otro antitrombótico, más potente que la HNF, al actuar sobre el factor IIa (efecto antitrombínico, especialmente sobre la antitrombina III), que determina su acción inhibidora de la agregación plaquetaria e inhibición y disgregación del trombo blanco plaquetario. Además, al bloquear el efecto de la trombina sobre el fibrinógeno, impide parcialmente la formación de fibrina, que constituye la malla que atrapa y consolida ambos tipos de trombos. No se conocen aún todos sus posibles mecanismos de acción, entre las cuales pudiera estar la activación del plasminógeno tisular (tPA), según observación que se valoró experimentalmente en 1987(34) o la inhibición del PAI-I, además de otras menos conocidas. De cualquier manera, no todas las HBPM son iguales en su mecanismo de acción, por lo que se justifican nuevas experiencias que nos permitan deducir el mecanismo de cada una de ellas.
Por las razones expuestas, iniciamos su uso en la cardiopatía isquémica descompensada o subaguda (1995), según nuestra clasificación propuesta (tabla 6) (41). En Marzo de 1995 coincidimos con investigaciones europeas que habían iniciado su efecto en la cardiopatía isquémica metabólica, en estudio multicéntrico (Frics), cuyos resultados fueron publicados en 1996(25). Con base a estudios anteriores en cuanto a la terapéutica dietético farmacológica causal(42,43), nuestra investigación al respecto de la HBPM fue publicada en 1998(44). En este estudio (clínico y electrocardiográfico, abierto, en 42 pacientes), se utilizó la dalteparina sódica a dosis de 5.000 UI diariamente durante 10 días, conjuntamente con indobufeno, a dosis de 400 mg diarios, que actúa como antiagregante plaquetario de acción exclusiva sobre los receptores plaquetarios de TXA2, y trimetazidina, a dosis de 60 mg diarios, cuya mecanismo de acción es ser cardioprotector del miocito isquémico, al cambiar la ruta metabólica intracelular de ácidos grasos a carbohidratos, con ahorro de energía y que además amortigua el efecto destructor de los RLO. Después de la aplicación de la HBPM se continuó con estos dos últimos productos por tres meses, cuando se valoró el resultado. El diagnóstico establecido en todos los casos fue el de cardiopatía isquémica metabólica subaguda, bien por presentar angina inestable (4 casos) o por tener este síntoma, asociado a alteraciones electrocardiográficas isquémicas, en 38 casos. Algunos de ellos presentaron también disnea, angustia y debilidad. La edad promedio fue de 64.2 años (38 - 82 años), con ligero predominio de hombres (28 casos) sobre mujeres (23 casos). El control fue ambulatorio en 41 de ellos y fue necesaria la hospitalización solamente en el paciente que además presentó desde su inicio taquicardia paroxística supraventricular. Ello significa que no obstante el cuadro subagudo, que para la mayoría amerita hospitalización, con el tratamiento propuesto se puede efectuar sin riesgo para el paciente de manera ambulatoria. Los resultados clínicos (dolor precordial) se exponen en la tabla 7 y los electrocardiográficos en las tablas 8,9,10,11. En las tablas 12,13 se comparan los resultados de nuestro estudio con el estudio Frics.
Tabla 4: Algunos estudios clínicos realizados hasta la fecha que demuestran las bondades de las Hbpm en la Cardiopatía Isquémica Metabólica.
| Estudio | n | Tipo de HBPM | Tiempo de administrc. | Resultados |
| Reducción del daño miocárdico con el tratamiento prolongado con HBPM en enfermedad coronaria inestable (24) | 1.276 | Dalteparina | 40 días | El tratamiento a largo plazo con HBPM disminuye el número de IM en pacientes con enfermedad coronaria inestable |
| FRISC II (26) | Dalteparina | 5-7 meses | Redujo muerte, reinfarto, revascularización y uso de HNF | |
| ESSENCE (27) | 3.171 | Enoxaparina | 14 días | Enoxaparina fue superior a la Heparina (p<0,001) en la reducción de eventos isquémicos en pacientes con angina inestable o infarto No- Q. |
| REDUCE (28) | 625 | Reviparina | 28 días | La administración de Reviparina no disminuye la reestenosis post-PCTA |
| FRIC S (25) | 1.482 | Dalteparina | 6-45 | Las HBPM son al menos tan efectivas como la HNF para reducir nuevos eventos isquémicos después del IM |
| TIMI 11A: Estudio sobre la presencia de complicaciones hemorrágicas durante el tratamiento con HNF comparada con enoxaparina (29) | 3.910 | Enoxaparina | 14 días | La enoxaparina provoco menos reacciones de tipo hemorrágicas que la heparina no fraccionada |
| TIMI 11B: Estudio sobre la eficacia en el uso de HNF comparada con enoxaparina en la angina inestable y el infarto No-Q (30,31) | 3.910 | Enoxaparina | Hasta 43 días redujo | El tratamiento con enoxaparina el 20 % en muertes y eventos isquémicos graves en comparación con la HNF. Este beneficio se mantuvo hasta el cierre del estudio (43 días). Este tratamiento no estuvo relacionado con un aumento de fenómenos hemorrágicos |
| Metanálisis sobre el uso de HBPM en enfermedad coronaria (32) | >6.000 | Enoxaparina Dalteparina | - | Metanálisis que demuestra reducción significativa a los 43 días de tratamiento en mortalidad e IM con HBPM comparado con HNF (7.1 vs. 8.6%, P = 0.02). |
Tabla 5: Cuadro clínico comparativo Heparina no fraccionada / Heparina Bajo PM.
| CONCEPTO | HEPARINA NO FRACCIONADA (HNF) | HEPARINA BAJO PESO MOLECULAR (HBPM) |
| PESO MOLECULAR | 13.000 D | 5.000 D |
| AFINIDAD POR AT III | + | ++++ |
| INTERACCIÓN CON FP4 (ANTIHEPARINA) | ++++ | + |
| AFINIDAD POR ENDOTELIO (METABOLIZACIÓN Y NEUTRALIZACION). | ++++ | + |
| INTERACCIÓN CON GLUCOPRO TEINAS RICAS EN HISTADINA (MENOR NEUTRALIZACION) | ++++ | + |
| VIDA MEDIA | + | ++++ |
| EFECTO ANTITROBÓTICO | + | ++++ |
| EFECTO ANTICOAGULANTE | ++ | ++ |
| AUM. ACTIV. TISULAR DEL PLASMINÓGENO | - | ++ |
| SE INACTIVA POR SULFATO DE PROTAMINA | SI | SI |
| CRUZA BARRERA PLACENTARIA | SI | SI |
| VÍA Y DOSIS HABITUAL | EV, 5.000 U SEGUIDO DE 1.000 U/H | SUBCUTANEA, 120 UI POR K / D |
| CONTROL DE LABORATORIO | SI | NO |
| HOSPITALIZACIÓN | SI | NO |
| USO EN TROMBOCITOPENIA | NO | SI |
| EFECTOS TÓXICOS : TERATOGEN. MUTÁGENOS | SI | NO |
| EFECTOS INDESEABLES A DOSIS HABITUAL (HEMORRAGIA, TROMBOCITOP, OSTEOPOROSIS) | SI | NO |
BERMÚDEZ–ARIAS (41)
Tabla 6: Clasificación clínico-Electrocardiográfica de la Cardiopatía Isquémico Metabólica (CIM)
| CONCEPTO | CLINICA | CLASE | ECG | CLASE |
| CRONICA | ANGINA ESTABLE (ULT. 3 MESES) | DOLOR PRECORDIAL LEVE, SIN CAMBIOS, ESCASA DURACIÓN | SIN ALTERACIONES ULTIMOS 3 MESES | *ST (LESIÓN) *T (ISQUEMIA) *QRS (NECROSIS) |
| SUBAGUDA | ANGINA INESTABLE (ULT. 3 MESES) A.PRIZMETAL | ITERATIVA S.INTERMED. (CIM SILENTE) A. INESTABLE INFARTO NO-Q | CAMBIOS RECIENTES CON ANGINA O SIN ELLA *ALT. QRS | *ALT. ST *ALT. T *TRAST. DEL RITMO *BLOQUEO DE RAMA IZQ. INFARTO NO-Q (DESN. NEG. ST; T NEG. PROFUND.) |
| AGUDA | IM AGUDO | DOLOR PRECORDIAL INTENSO, DURACIÓN PROLONGADA | CAMBIOS MARCADOS | *IM"Q" (ST+T+QRS) |
BERMÚDEZ–ARIAS (41)
Tabla 7: Resultados. Dolor Precordial
| INTENSIDAD | CASOS | % | MEJORO | MEJORO | MEJORO | NO MEJORO |
| 1º SEMANA | (42) | 100% | 75% | 50% | ||
| REPOSO/PEQUEÑOS ESFUERZOS | 27 | 64.2 | 55.5% | 14.8% | 29.6% | 0 |
| MODERADOS ESFUERZOS | 13 | 30.9 | 84.6% | 15.3% | 0 | 0 |
| GRANDES ESFUERZOS | 2 | 4.7 | 100% | 0 | 0 | 0 |
| NO | - | - | - | - | - | - |
Tabla 8: Resultados. Mejoría ECG-12D
| A LOS 7 días | A LOS 15 DÍAS | A LOS 3 MESES |
| 25 CASOS (59.5 %) | 10 CASOS (23.8 %) | 7 CASOS (16.6 %) |
Tabla 9: Resultados. Frecuencia cardíaca.
| 2-12 SEM | CASOS (42) | % | AUM | % | DISM | % | IGUAL | % |
| TS EXTR. >130 | 1 | 2.3 | - | - | 1 | 100 | - | - |
| TS MODERADA 111-130 | 0 | 0 | - | - | - | - | - | - |
| TS 91-110 | 12 | 28.5 | - | - | 12 | 100 | - | - |
| NORMAL 70-90 | 19 | 45.2 | - | - | 3 | 15.7 | 16 | 84.2 |
| BS 50-69 | 9 | 21.4 | 6 | 66.6 | - | - | 3 | 33.3 |
| BS MODERADA 40-49 | 1 | 2.3 | 100 | - | - | - | - | - |
| BS EXTR. <39 | 0 | 0 | - | - | - | - | - | - |
Tabla 10: Resultados. Arritmias y bloqueos
| ARRITMIAS, BLOQUEOS 2-12 SEM | CASOS (42) | % | MEJORIA (CASOS) | % EFECTIVIDAD |
| EXTRAS. VENT | 4 | 9.5 | 3 | 75 |
| EXTRAS. AURIC | 1 | 2.3 | 1 | 100 |
| TAQ.PARX.AUR | 1 | 2.3 | 1 | 100 |
| TAQ. NODAL | 1 | 2.3 | 0 | 0 |
| FIBRILAC. AUR. | 2 | 4.6 | 0 | 0 |
| BLOQUEO RD | 2 | 4.6 | 1 | 50 |
| BLOQUEO RI | 1 | 2.3 | 0 | 0 |
| HEMIBLOQUEO ANTEROSUPERIOR | 2 | 4.6 | 0 | 0 |
Tabla 11: Resultados de las Alteraciones ECG Isquémicas Directas
| 2 A 12 SEMANAS | 49 ALTERACIONES | % | 42 CASOS | - | - | - |
| MEJORÍA 100% | MEJORÍA 99-50% | MEJORÍA <50% | NO MEJORÓ | |||
| ZONA INACTIVABLE CORONARIA DER. | 6 | 12. 2 | 4 CASOS 9. 5 % | 1 CASO 2. 3 % | 0 | 1 CASO 2. 3 % |
| ZONA INACTIVABLE CORONARIA IZQ. | 7 | 14. 2 | 6 CASOS 14. 2 % | 0 | 0 | 1 CASO2 . 3 % |
| ST-T CORONARIA DERECHA | 5 | 10. 2 | 5 CASOS 11. 9% | 2 CASOS 4. 6 % | 2 CASOS 4. 6 % | 0 |
| ST-T CORONARIA IZQ. | 31 | 63. 2 | 11 CASOS 26. 1% | 7 CASOS 16. 6 % | 1 CASO 2.3% | 1 CASO 2.3% |
| TOTAL | 49 ALTER. | 100 | - | - | - |
Tabla 12: Análisis comparativo de los resultados con Dalteparina Sódica en los estudio Frics (Europa, 1996) y el nuestro (Maracaibo, 1996)
| CONCEPTO | FRICS, N: 1506 | BERMUDEZ, N: 42 |
| TIPO DE ESTUDIO | CONTROLADO | ABIERTO |
| METODOS | CLINICO / ECG / CINE CORONARIOGRAFÍA. | CLINICO / ECG |
| DIAGNOSTICO | NO SE PRECISA | CIM SUBAGUDA |
| DROGAS | FRAGMIN + ASPIRINA | FRAGMIN + INDOBUFENO + TRIMETAZIDINA + POTASIO |
| CONTROL | HOSPITALIZACIÓN / AMBULATORIO | AMBULATORIO |
| DOSIS | 10.000 UI POR 6 DIAS+7.500 UI POR 35 A 45 DIAS MAS | 5.000 UI DIARIAS POR 10 DIAS |
| MUERTES (FRAGMIN/PLACEBO) | 7 / 8 (NO SIG: 0.8) | NO HUBO (ALTAM. SIG) |
| NUEVOS IM | 10 / 33 (SIG: 0.001) | NO HUBO (ALTAM. SIG) |
| REVASCULARIZACION | 3 / 9 (NO SIG: 0.07) | NO HUBO (ALTAM. SIG) |
| HEPARINIZACION | 26 / 58 (SIG: 0.001) | NO HUBO (ALTAM. SIG) |
Nota: La diferencia a favor de nuestro estudio con relación al estudio Frics, puede atribuirse a la diferencia en el número de casos y al resto del tratamiento que acompañó a la HBPM, que en el primer caso se acompaño de indobufeno, trimetazidina y potasio, mientras que en el segundo (estudio Frics) se hizo con aspirina, que actúa como antiagregante plaquetario al bloquear la ciclooxigenasa 2 y por ello disminuir la acción del TXA2, pero que por la misma razón también disminuye la prostaciclina, efecto que no solamente aumenta la trombosis sino que también produce vasoconstricción. A favor de nuestro estudio estuvo la dosis significativamente inferior de HBPM
Tabla 13: Estudio clínico comparativo de tres estudios
| HBPM | HOSPITA- LIZACION % | MORTA- LIDAD % | NECESIDAD DE HNF % | REVASCU- LARIZACIÓN % | REIN- FARTO% | ANGINA RECURRENTE % | TOTAL EVENTOS % |
| ESSENCE | |||||||
| ENXOAPARINA + ASPIRINA. CORTE A LOS 14 DÍAS | ¿? / 3180 | 1.7 | ¿? | ¿? | 5.9 | 17 | 23.6 |
| FRICS II | |||||||
| DALTEPARINA+ ASPIRINA. CORTE A LOS 5-7 MESES | TODOS 1506 | 1.8 vs 4.8 | 3.8 vs 7.7 | 0.4 vs 1.2 | ¿? | ¿? | 5.4 vs 10.3 |
| BERMÚDEZ-ARIAS | |||||||
| DALTEPARINA+ INDOBUFENO+ TRIMETAZIDINA + POTASIO. CORTE A LOS 3 MESES | 1/ 42 | 0 | 0 | 0 | 0 | 0 | 0 |
Tabla 14: Factores Naturales Protectores Contra la Trombosis
| * PGI2 O PROSTACICLINA: |
| PROSTAGLANDINA DERIVADA DEL ENDOTELIO, DEPENDIENTE DE LA ACCION DE LA CICLOOXIGENASA SOBRE EL ACIDO ARAQUIDONICO. (EL AAS INHIBE SU FORMACION). NO ES AFECTADO POR LOS RADICALES LIBRES DE OXIGENO. ESTIMULA ADENILCICLASA Y AUMENTA AMPc , Y ACTUA COMO ANTIAGREGANTE PLAQUETARIO Y VASODILATADOR |
| * ANTITROMBINA (ATT) III: |
| INTERFIERE POR VIA INTRINSECA Y POR VIA COMUN LA COAGULACION. AMERITA DE CELULAS ENDOTELIALES INTACTAS PARA SU ACCION. ACTUA CONTRA LA ARGININA PARA FAVORECER LA UNION DE LA TROMBINA CON LA FIBRINA, LO CUAL INACTIVA LA PRIMERA COMO FACTOR DE COAGULACION Y TROMBOSIS. |
| * PROTEOGLICANO DE HEPARAN SULFATO: |
| AUMENTA ACCION DE LA ATT III. |
Tabla 15: Fibrinógeno y Factor VIII de la Coagulación
| * NO SON AFECTADOS POR LA ATT III. |
| * AL NO SER INACTIVADAS POR ATT III, SUS PEQUEÑOS AUMENTOS PRODUCEN GRAN HIPERCOAGULABILIDAD, ESPECIALMENTE SI ES POR RUPTURA DE PLACA, CON AUMENTO DEL FACTOR TISULAR DE LA COAGULACION Y LA TROMBOSIS (TROMBOPLASTINA) |
| * ESTÁN MAS RELACIONADOS CON ENFERMEDAD TROMBÓTICA CORONARIA. |
| * AMBOS AUMENTAN CON EDAD, OBESIDAD, CONTRACEPTIVOS, OBESIDAD, MENOPAUSIA, DIABETES, TABAQUISMO. |
| * AMBOS SON DISMINUIDOS POR DIETA VEGETARIANA NO LACTEA. |
| * LAS LIPOPROTEINAS ACTIVAN EL FACTOR VIII. |
| * LA MAYOR CANTIDAD DE FIBRINÓGENO PRODUCE UNA MAYOR CANTIDAD DE FIBRINA, QUE LA HACE MAS RESISTENTE A LA LISIS PRODUCIDA POR LA PLASMINA. |
| * EL FIBRINÓGENO AUMENTA EL ENLACE DE LOS RECEPTORES PLAQUETARIOS CON EL FACTOR VIII, LO CUAL AUMENTA LA AGREGACION PLAQUETARIA. |
RESULTADOS
Al agregar la HBPM al tratamiento dietético farmacológico causal que ya habíamos iniciado varios años antes, notamos la mejoría clínica y electrocardiográfica en menor tiempo que el que se había logrado con las otras medidas (indobufeno, trimetazidina y potasio). Fisiológicamente, la cascada de la agregación plaquetaria, se inicia cuando se despule el endotelio y se descubre el colágeno IV de la íntima o capa subendotelial de la arteria. El ADP y el factor VIII o de von Willebrand actúan como activadores de los receptores Ia-Ib y IIb-IIIa plaquetarios, iniciando la adhesividad de la primera capa de plaquetas. Una vez adheridas las plaquetas ellas se activan, mediante la liberación de calcio, activación de la proteinkinasa C y del sistema actino-miosina intraplaquetario, liberando el contenido de los gránulos densos y de los gránulos alfa, sustancias que a su vez se fijarán en los receptores respectivos (IIb/ IIIa, TXA2, serotonina, trombina, adrenalina, factor de agregación plaquetaria o factor VIII, ADP, factor VIIa y factor de crecimiento de células musculares lisas derivado de plaquetas o PDGF) de las otras plaquetas para que se continúe la agregación plaquetaria. El PDGF no actúa directamente agregando plaquetas sino que lo hace como factor quimiotáctico, proliferativo e hipertrófico de CML de la capa media arterial, y en consecuencia como vasoconstrictor y aterogénico directo, razones por las cuales lo consideramos como el elemento de enlace entre la trombosis actual y la mayor aterosclerosis futura. Dentro de los antiagregantes plaquetarios, esta sustancia solamente es inhibida por el indobufeno y posiblemente también por las HBPM.
El manejo simple de los pacientes, mediante clínica y electrocardiografía (sin hospitalización, aun cuando se encuentren inestables), pero aplicando los medicamentos de acuerdo a su mecanismo de acción con criterio fisiológico y bioquímico molecular, ha proporcionado los resultados extraordinarios de este método de tratamiento exclusivamente no invasivo y únicamente basado en lo que hemos denominado, por sus resultados, "trombectomía química". Ello nos permite plantearlo como una magnífica propuesta, más que como una alternativa en el tratamiento de la CIM (Fig. 3). Una vez restaurado el flujo en mayor o menor cantidad, resta defender la célula miocárdica de la acción de los RLO, lo cual logramos con la trimetazidina, con lo cual se evitan nuevas agresiones a las membranas celular y mitocondrial, y las arritmias de reperfusión, por el así denominado "efecto paradójico del oxígeno". De cualquier manera, se observó en la mayoría de los pacientes estabilización de la frecuencia cardiaca (FC) en cifras cercanas a las normales (70 a 90 LPM), es decir, permanencia en cifras normales, cuando así era antes de instalar el tratamiento, disminución en la taquicardia sinusal y aumento en la bradicardia sinusal con el efecto único del tratamiento antiisquémico propuesto. Este hallazgo es de gran valor, desde que se sabe por el estudio de Framingham el aumento significativo de la mortalidad tanto en hombres como en mujeres hipertensas cuando la FC se acelera, llegando a duplicarla cuando alcanza más de 85 latidos por minuto si se compara con la de 65 por minuto.
3.A.1 ECG- D previo al tratamiento. Paciente masculino de 57 años con dolor precordial intenso que había iniciado 48 horas de la consulta. Con cardiopatía isquémica metabólica aguda por IM en fase aguda septal medio y bajo (QS en V1 V2 y v3) más lesión e isquemia subepicárdica en las mismas derivaciones y además en V4, V5, V6, en D2, D3, A VF Y EN D1 Y a VL, correspondientes a la zona septal media, septal baja y lateral baja, a la cara inferior y a la zona lateral alta del ventrículo izquierdo respectivamente. Se inició tratamiento con dieta cronobiológica, antioxidante, polarizante (CAP), y farmacológico con dalteparina (HBPM) 5000 UI por 10 días, indofubeno (400 mg diarios), trimetazidina (60 mg diarios) y solución polarizante continúa durante los cinco días de hospitalización. La solución polarizante se omitió al ser dado de alta y la HBPM se continuó hasta el décimo día. La trimetazidina y el indofubeno se mantuvieron en las mismas dosis.
3.A.2 El paciente se controló cada semana. A los quince días de haber iniciado el tratamiento dietético farmacológico causal, además de haber desaparecido el dolor a las 24 horas, se observa mejoría del trazado electrocardigráfico caracterizado por desaparición de la zona eléctricamente inactivable septal media y baja (recuperaciópn de la R en V1, V2 y V3) y desaparición de la lesión subepicárdica en todas las derivaciones en las cuales se observaba a expensas del aumento de la isquemia subepicárdica.
3.A.3 Un mes después el trazado electrocardiográfico se ha normalizado totalmente.
3.A.4 Cinco meses más tarde se mantiene la normalidad del paciente y del ECG-12D. A partir de esta fecha se disminuyó la dosis de indofubeno a 300 mg diarios y se mantuvo la misma dosis de trimetazidina.
3.B.1 Paciente masculino de 55 años, que mantiene dolor desde 3 días antes del registro del ECG-12D que se muestra en la figura. Se observa el infarto del miocardio en fase aguda localizado en cara inferior. Por falta de recursos económicos se inició ambulatoriamente el tratamiento dietético farmacológico causal con dieta CAP y farmacológico con enoxaparina (HBPM) a dosis de 60 mg diarios). El tratamiento se continuó con indofubeno y trimetazidina.
3.B.2 El dolor en reposo desapereció a las 24 horas. Una semana después se observa extensión el infarto a las regiones septal media, septal baja, y lateral baja del ventrículo izquierdo 9territorio de la descendente anterior posdiagonal) Sin embargo, se observa desaparición de la lesión subepicárdica en D2, D3 y a VF con aumento de la isquemia subepicárdica, lo cual se interpreta como inicio de mejoría de la irrigación en la cara inferior. El ECg-12D se mantuvo igual hasta un mes después. Se mantuvo el mismo esquema de tratamiento.
3B.3 A los dos meses el paciente se siente asintomático. En el ECG-D se observa desaparición de la zona eléctricamente inactivable de las regiones septal media y baja y en la lateral baja del ventrículo- izquierdo. En cara inferior, región de la coronaria derecha, la mejoría es solo parcial, manifestada por aumento de la R en D2, si se compara con la R en la misma derivación de los trazados anteriores. La isquemia subepicárdica disminuyó en las derivaciones en las cuales de manifestaba.
3B.4 Siete meses más tarde, habiéndose mantenido el mismo esquema de tratamiento, se observa normalización del trazado con excepción de la zona fibrótica (QS) en D3 y a VF.
Quedan aun por discutir los mecanismos bajo los cuales este tratamiento logra llegar hasta la desaparición de los signos de necrosis en un número significativo de casos (9 de 42), es decir, en el 22 por ciento de ellos, lo cual es posible mediante la hipótesis que muchas de estas células se encuentran en estado de hibernación (en etapa de cronicidad) o en estado de atontamiento (en etapa aguda) y no de fibrosis. O sea, que la célula hibernada se encuentra como si estuviera en estado fetal, lo cual significa que están inactivas funcionalmente, pero aún viables. La célula muere cuando el ATP llega y se mantiene en cero. En ese caso, el Ca++ intracelular activa la fosfolipasa A2, que degrada los lípidos de la membrana y permite el escape del ácido glutámico, el cual se une con los receptores n-metil d-aspartato (cascada del gluconato). Al suceder ello se abren sin control y definitivamente los canales de calcio, electrólito que entonces inunda la célula, activa las enzimas lisosómicas (proteolíticas) y destruye y provoca la muerte celular. Sin embargo, en el sector anatómico correspondiente al segmento electrocardiográfico inactivable, aparente e irreversiblemente muerto, es posible que haya algunas moléculas de ATP y algo de oxígeno, en cuyo caso la cascada del glutanato y la muerte celular no es definitiva en forma total, quedando algunas células hibernadas (en período postisquémico) y otras atontadas, ambas funcionalmente disminuidas, aunque viables en caso de restablecimiento del flujo arterial coronario(41).
Nueve de nuestros casos mostraron desaparición total de los signos de isquemia, lesión y necrosis, lo cual se observó en su mayor proporción en la paciente que luego de haber sufrido un infarto del miocardio inferior (coronaria derecha) y septal medio y bajo extendido a pared lateral baja del ventrículo izquierdo (coronaria izquierda), se obtuvo una total mejoría, que sólo se explicaría bajo la presencia de un posible efecto trombolítico por activación del plasminógeno tisular, tal como ha sido encontrado experimentalmente con las HBPM(34). Estos resultados inmensamente favorables y persistentes en nuestra casuística si se comparan con los de angioplastia, la cirugía de revascularización y otras intervenciones mecánicas propuestas y esquemas de tratamiento médico, nos permiten plantear a la consideración de la cardiología, este procedimiento terapéutico ("revascularización farmacológica" por intermedio de la "trombectomía química y protección miocárdica") como la mejor alternativa actual para el tratamiento de la cardiopatía isquémica. A estos resultados debe agregarse además, la escasa aparición de efectos indeseables menores y la ausencia de otros de mayor intensidad y peligrosidad para la vida de los pacientes.
Los últimos estudios con HBPM con mayor número de casos reportados han sido el estudio ESSENCE(27), en el cual se evaluó la eficacia de la enoxaparina (Clexane) en la angina inestable y el infarto miocárdico No-Q, a doble ciego, randomizado, controlado con placebo y en grupo paralelo con HNF, multicéntrico. El número de pacientes fue 3180, edad igual o mayor de 18 años (media de 64 años), 33% mujeres, 46% con infarto premio. El seguimiento fue de 30 días. El tratamiento se hizo con Enoxaparina 1 mg/kg sc cada 12 horas, o HNF 5000 UI intravenoso seguido de infusión continua de para mantener el PTT al doble del control, o placebo. Terapia concomitante: aspirina 100-325 mg diarios. El resultado preliminar en 3019 pacientes indicó: El porcentaje total de eventos a los 14 días arrojó las cifras siguientes: mortalidad de 1.7%, reinfarto subsecuente de 5.9% y angina recurrente de 17%, con una rata total de eventos del 23.6%.
El estudio FRICS II (26): fue un estudio de 1506 pacientes con edad media de 71 años (963 hombres), hospitalizados 72 horas antes de iniciar el tratamiento, randomizado, doble ciego, controlado con placebo y en grupo paralelo, con seguimiento entre 5 y 7 meses. Se empleó la dalteparina (HBPM) durante las crisis de inestabilidad en la enfermedad arterial coronaria, a dosis de 120 U/kg (máximo 10.000) cada 12 horas los primeros 6 días, seguido de 7500 U diarias por los siguientes 35 a 45 días, o placebo. Se agregó aspirina 300 mg inicialmente, seguido de 75 mg diarios, y otros medicamentos como antagonistas del calcio, betabloqueantes, nitratos orgánicos y nitroglicerina cuando fue necesario. La mortalidad por infarto en los primeros 6 días fue 1.8% contra 4.8 por ciento; necesidad de HNF 3.8% contra 7.7%, y de revascularización 0.4% contra 1.2 %. La diferencia total, incluyendo muertes, IM, revascularización y HNF, en conjunto fue de 5.4% contra 10.3% (0.52: 0.37-0-75). La diferencia en muertes y reinfarto se mantuvo a los 40 días. El efecto sobre la reactivación del proceso y el reinfarto se produjo con la disminución de las dosis y fue más pronunciado en los fumadores. 4 a 5 meses después de finalizar el tratamiento no hubo diferencias significativas en la mortalidad, reinfarto o revascularización. El régimen fue seguro y las quejas fueron adecuadas.
Una comparación de los resultados de los dos estudios anteriores con el nuestro se expone en la tabla 9.
REFERENCIAS BIBLIOGRAFICAS
1. Schafer AI. Pathophysiology of thrombosis. In: Loscalzo J, et al. (eds). Vascular Medicine: A Textbook of Vascular Biology and Disease. Boston: Little Brown & Co., 1992: pp. 307-333. [ Links ]
2. Klein W, Buchwald A, Hillis SE, et al. Comparison of low-molecular-weight heparin with unfractionated heparin acutely and with placebo for 6 weeks in the management of unstable coronary artery disease. Fragmin in Unstable Coronary Artery Disease Study (FRIC). Circulation 1997;96:61-68. [ Links ]
3. Yun DD, Alpert JS. Acute coronary syndromes. Cardiology 1997;88:223-237. [ Links ]
4. Becker RC. Antiplatelet therapy. Science & Medicine 1996;(July/August):12-21. [ Links ]
5. Olson St, Björk I. Regulation of thrombin by antithrombin and heparin cotactor II. In: Berliner, LJ (ed). Thrombin: Strucuture and Function. New York: Plenum Press, 1992: pp. 159-217. [ Links ]
6. Rosenberg RD. The heparin-antithrombin system: A natural anticoagulant mechanism. In: Colman RW, Hirsh J, Marder VJ, Salzman EW (eds). Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 2nd edition. Philadelphia: JB Lippincott, 1987: pp. 1373-1392. [ Links ]
7. Hirsh J. Heparin. N Engl J Med 1991;324:1565-1574. [ Links ]
8. Instability in Coronary Artery Disease (FRISC) Study Group. Low molecular weight heparin during instability in coronary artery disease. Lancet 1996;347:561-568. [ Links ]
9. Theroux P, Waters D, Lam J, et al. Reactivation of unstable angina after the discontinuation of heparin. N Engl J Med 1992;327:141-145. [ Links ]
10. Stein B, Fuster V . Pharmacology of anticoagulants and platelet inhibitor drugs. In: Schlant RC, Alexander RW (eds). Hurst’s The Heart, Volume I, 8th edition. New York: McGraw-Hill, 1994: pp. 1309-1326. [ Links ]
11. Antiplatelet Trialist Collaboration. Collaborative overview of randomized trials on antiplatelet therapy: I. Prevention of death, myocardial infarction and stroke by antiplatelet therapy in various categories of patients. BMJ 1994;308:81-106. [ Links ]
12. Campbell RWF, Wallentin L, Verheught FWA, et al. Management strategies for a better outcome in unstable coronary artery disease. Clin Cardiol 1998;21:314-322. [ Links ]
13. Markwardt F. Development of hirudin as an antithrombotic agent. Semin Thromb Hemost 1989;15:269-282. [ Links ]
14. Harker LA, Fuster V. Pharmacology of platelet inhibitors. J Am Coll Cardiol 1986;8(Suppl 1B):21B-32B. [ Links ]
15. Schrör K. Antiplatelet drugs: A comparative review. Drugs 1995;50:7-28. [ Links ]
16. Jang IK, Gold HK, Ziskind AA, et al. Prevention of platelet-rich arterial thrombosis by selective thrombin inhibition. Circulation 1990;81:219-225. [ Links ]
17. Sarembock IJ, Gertz SD, Gimple LW, et al. Effectiveness of recombinant desulphatohirudin in reducing restenosis after balloon angioplasty of atherosclerotic femoral arteries in rabbits. Circulation 1991;84:232-243. [ Links ]
18. Bittl JA, Strony J, Brinker JA, et al. Treatment with bivalirudin (Hirulog) as compared with heparin during coronary angioplasty for unstable or postinfarction angina. N Engl J Med 1995;333:764-769. [ Links ]
19. Fuchs J, Cannon CP, the TIMI 7 Investigators. Hirulog in the treatment of unstable angina. Results of the Thrombin Inhibition in Myocardial Ischemia (TIMI) 7 Trial. Circulation 1995;92:727-733. [ Links ]
20. Heras M, Chesebro JH, Penny WJ, et al. Effects of thrombin inhibition on the development of acute thrombus deposition during angioplasty in pigs: Heparin versus recombinant hirudin, a specific thrombin inhibitor. Circulation 1989;79:657-665. [ Links ]
21. White HD, Aylward PE, Frey MJ, et al. Randomized, double-blind comparison of hirulog versus heparin in patients receiving streptokinase and aspirin for acute myocardial infarction (HERO). Circulation 1997;96:2155-2161. [ Links ]
22. Majerus PW, Broze GJ, Miletich JP, Tollefsen DM. Anticoagulant, thrombolytic, and antiplatelet drugs. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 9th edition. New York: McGraw-Hill, 1996: pp. 1341-1359. [ Links ]
23. Théroux P, Ouimet H, McCans J, et al. Aspirin, heparin or both to treat acute unstable angina. N Engl J Med 1988;319:1105-1111. [ Links ]
24. The GUSTO IIa Investigators. Randomized trial of intravenous heparin versus recombinant hirudin for acute coronary syndromes. Circulation 1994;90:1631-1637. [ Links ]
25. Study group: Fragmin during instabillity in coronary artery disease (FRICS STUDY). Lancet 1996; 347: 561-568. [ Links ]
26. Wallentin L et study group: Fragmin and Fast Revascularistion during in Stability in Coronary artery disease (FRISC II STUDY). Lancet: 1999; 354: 708-715. [ Links ]
27. Cohen M, Blaber R, Demers C, Gsurfinkel EP, Langer A, Fromell G, Turpie AGG, Premmereur J: Efficacy and Safety of subcutaneous Enoxaparin in unstable angina and Non-Q myocardial infarction. Ongoing trial (ESSENCE STUDY). J Thromb Thrombolyusis 1997; 4: 271-74 [ Links ]
28. Preisack MB; Baildon R; Eschenfelder V; Foley D; Garcia E; Kaltenbach M; Meisner C; Selbmann HK; Serruys PW; Shiu MF; Sujatta M; Bonan R; Karsch KR: Low molecular weight heparin, reviparin, after PTCA: results of a randomized double-blind, standard heparin and placebo controlled multicenter study (REDUCE STUDY]. Z Kardiol 1997;86(8):581-91 [ Links ]
29. Turpie AG : Management of acute coronary syndromes with low molecular weight heparin: TIMI 11A and 11B. Can J Cardiol 1998;14 Suppl E:20E-23E. [ Links ]
30. TIMI 11B. Enoxaparin versus unfractionated heparin for unstable angina or non-Q-wave myocardial infarction: a double-blind, placebo-controlled, parallel-group, multicenter trial. Rationale, study design, and methods. Thrombolysis in Myocardial Infarction (TIMI) 11B Trial Investigators. Am Heart J 1998;135 (3):S353-60. [ Links ]
31. Assessment of the treatment effect of enoxaparin for unstable angina/non-Q-wave myocardial infarction. TIMI 11B-ESSENCE meta-analysis. Circulation 1999; 100 (15):1602-8. [ Links ]
32. Assessment of the treatment effect of enoxaparin for unstable angina/non-Q-wave myocardial infarction. TIMI 11B-ESSENCE meta-analysis. Circulation 1999;100(15):1602-8. [ Links ]
33. Holm HA et al: Subcutaneous heparin treatment of deep venous thrombosis: A comparison of unfractionated and low molecular weight heparin. Haemostasis 1986, 16:30-37. [ Links ]
34. Janvier G et al: Treatment of deep venous thrombosis with a very low molecular weight triglyceride lipase (HTGL). Haemostasis 1987, 7: 4958-4966. [ Links ]
35. Pérez Resquejo JL: Hematología para cardiólogos. Editexto. Caracas, 1991. Cita: Thrombos Hemos 1987, 58: 947. [ Links ]
36. Borel JP et al: Bioquímica Dinámica. Editorial Médica Panamericana. Buenos Aires, 1989. [ Links ]
37. Goodman Gilman A, Rall TW, Nies AS, Taylor P: Las bases farmacológicas de la terapéutica. Novena Edición. McGraw-Hill, 1996. [ Links ]
38. Kanel WB, McGee D y Gordon TA: A general cardiovascular risk profile. Am. J. Cardiol. 1976; 38: 46. [ Links ]
39. Opie LH: Stunning, hibernation, and calcium in myocardial ischemia and reperfusion. Kluwer Academic Publishers. Boston, 1992. [ Links ]
40. Harenberg, J: Pharmacology of low molecular weight heparins. Seminars in Thrombosis and hemostasis. 1990; 16: 12-17. [ Links ]
41. Bermúdez-Arias F: Electrocadiografía Diagnóstica. Editorial McGraw-Hill Interamericana de Venezuela. 1998. [ Links ]
42. Bermúdez-Arias F, Valbuena R, Soto L, Morillo Z: Efecto del indobufeno en la cardiopatía isquémica metabólica. Segundas Jornadas Occidentales de Cardiología. Maracaibo, 1996. [ Links ]
43. Bermúdez-Arias F, Baptista-Arrieta G, Fernández A: Firs study of trimetazidina (FAST STUDY). Arch Ven Farmcol y Terap 1995; 14: 39-53. [ Links ]
44. Bermúdez-Arias F, Chacín Chacín A, Bermúdez Pirela V y Cano Ponce C: Heparinas de bajo peso molecular en la cardiopatía isquémico metabólica. Resultados en 42 casos. Arch Ven Farmcol y Terap 1998; 17 (1): 50-62. [ Links ]
{kind=link}